Синдром Пфайффера, наблюдаемый у каждого из 100 000 новорожденных, связан с мутацией генов FGFR1 и FGFR2; Задача обоих этих генов - регулировать слияние черепных швов и развитие пальцев рук и ног.
Для диагностики синдрома Пфайффера основополагающими являются физикальное обследование, анамнез, рентгенологическая оценка черепа и пальцев рук и ног и, наконец, генетический тест.
В настоящее время люди, страдающие синдромом Пфайффера, могут рассчитывать только на симптоматическое лечение, то есть на то, что облегчает симптомы.
Краткий обзор черепных швов и их сращивания
Черепные швы - это фиброзные суставы, которые служат для соединения костей свода черепа (то есть лобных, височных, теменных и затылочных костей).
В нормальных условиях процесс сращивания черепных швов происходит в послеродовой период, начиная с 1-2 лет для одних элементов сустава и заканчивая в 20 лет для других. Этот длительный и ритмичный процесс слияния позволяет мозгу адекватно расти и развиваться.
- Наличие аномально больших и искривленных больших пальцев стопы так, что кажется, что они отодвигаются от других пальцев стопы (медиальное отклонение).
Таким образом, синдром Пфайффера является генетическим заболеванием, которое у носителей этого заболевания в основном определяет аномалии черепа и рук.
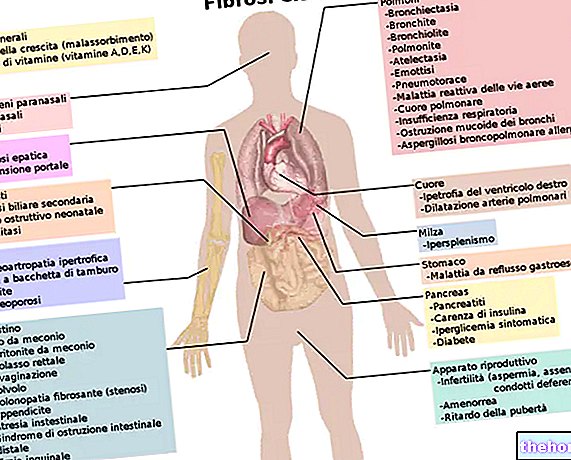
Однако, поскольку у читателей будет возможность узнать больше в главе, посвященной симптомам, синдром Пфайффера может быть связан с другими проблемами и другими физическими пороками развития.
Эпидемиология: насколько распространен синдром Пфайффера?
По статистике, каждый 100000 человек рождается с синдромом Пфайффера.
Вы знали, что ...
Генетических заболеваний, которые, как и синдром Пфайффера, вызывают краниосиностоз, около 150.
Среди них, помимо синдрома Пфайффера, важное значение имеют синдром Крузона, синдром Аперта и синдром Сэтре-Чотцена.
Что вызывает мутацию гена, связанную с синдромом Пфайффера?
Посылка: гены, присутствующие в хромосомах человека, представляют собой последовательности ДНК, задачей которых является производство основных белков в биологических процессах, необходимых для жизни, включая рост и репликацию клеток.
Когда они свободны от мутаций (то есть у здорового человека), гены FGFR1 и FGFR2 продуцируют в нужных количествах соответственно рецептор 1 фактора роста фибробластов и рецептор 2 фактора роста фибробластов, которые являются двумя рецепторными белками, необходимыми для маркировки выбор времени для сращивания черепных швов и для регулирования развития пальцев рук и ног (другими словами, они сигнализируют, когда наступает подходящее время для сращивания черепных швов, и контролируют формирование пальцев и стоп).
С другой стороны, когда они претерпевают мутации, наблюдаемые при синдроме Пфайффера, гены FGFR1 и FGFR2 становятся гиперактивными и производят вышеупомянутые рецепторные белки в таких огромных количествах, что время слияния черепных швов изменяется (они быстрее ) и процесс тренировки пальцев рук и ног происходит неправильно.
Синдром Пфайффера - аутосомно-доминантное заболевание.
Понять...
Каждый человеческий ген присутствует в двух копиях, называемых аллелями, одна из которых имеет материнское происхождение, а другая - отцовского происхождения.
Синдром Пфайффера имеет все характеристики аутосомно-доминантного заболевания.
Генетическое заболевание является аутосомно-доминантным, когда мутации одной копии гена, вызывающей его, достаточно, чтобы проявиться.
Типы синдрома Пфайффера
В 1993 году, после многочисленных исследований синдрома Пфайффера, американский врач Майкл Коэн опубликовал типологическую классификацию рассматриваемого генетического заболевания, в которой предсказывалось существование трех патологических вариантов, идентифицируемых просто с помощью терминов «Тип I», «Тип II» и Тип III »и все они имеют краниосиностоз и аномалии большого пальца стопы. Научно-медицинское сообщество немедленно приняло эту классификацию, и с тех пор эксперты по синдрому Пфайффера использовали ее в качестве диагностического инструмента и для оценки тяжести имеющегося генетического состояния; Фактически, следует отметить, что классификация доктора Коэна различает синдром Пфайффера на основе тяжести черепных и пальцевых аномалий, а также наличия других симптомов и признаков.
Обращаясь к деталям отдельных патологических вариантов, на данном этапе статьи важно подчеркнуть, что:
- В Тип I. это менее тяжелая версия синдрома Пфайффера, поскольку краниостеноз и аномалии большого пальца стопы имеют ограниченные последствия.
Другая важная информация: это связано с мутацией FGFR2, иногда в сочетании с мутацией FGFR1; это может быть наследственное или приобретенное состояние. - В Тип II это самая тяжелая версия синдрома Пфайффера, поскольку она связана с тяжелым краниосиностозом, почти несовместимым с жизнью, и с глубокими аномалиями в руках и ногах.
Другая важная информация: это происходит исключительно из-за мутации FGFR2; это всегда приобретенное состояние. - В Тип III это версия синдрома Пфайффера, которая по шкале тяжести находится чуть ниже типа II, но намного выше типа I, поскольку нынешний краниосиностоз почти такой же тяжелый, как и вариант, описанный в предыдущем пункте.
Другая важная информация: это происходит исключительно из-за мутации FGFR2; это всегда приобретенное состояние.
Краниоостеноз
У носителей синдрома Пфайффера краниосиностоз может, в зависимости от количества черепных швов, вовлеченных в процесс раннего сращения, иметь следующие последствия:
- Совершенно ненормальное вертикальное развитие головы в сочетании с недостаточным боковым расширением черепа. Следовательно, у пациента с синдромом Пфайффера длинная узкая голова;
- Формирование высокого и выпуклого лба;

- Повышенное внутричерепное давление, от которого зависят такие симптомы, как постоянная головная боль, проблемы со зрением, рвота, раздражительность, проблемы со слухом, проблемы с дыханием, изменения психического статуса, отек диска зрительного нерва;
- Интеллектуальный дефицит, ведущий к снижению IQ. Интеллектуальная недостаточность является результатом ограниченного пространства для роста, которым обладает мозг после преждевременного сращивания коронарных черепных швов;
- Отсутствие развития промежуточной части лица, которая кажется плоской, если не вогнутой;
- Наличие выпученных (проптоз), широко открытых и аномально расставленных глаз (глазной гипертелоризм);
- Наличие клювого носа;
- Неспособность развить челюсть (гипоплазия верхней челюсти), что приводит к скученности зубов;
- Клеверный вид головы («клеверный череп»). «Клеверный череп» вызывает гидроцефалию.
ТИП I
Синдром Пфайффера I типа связан с легким клиническим краниосиностозом, который очень часто ограничивается приданием черепу удлиненной формы, вызывающим заметно высокий лоб и плоское лицо.
При правильном лечении люди с синдромом Пфайффера I типа обычно ведут нормальный образ жизни и имеют нормальный IQ.
ТИП II
Синдром Пфайффера II типа - единственный патологический вариант, вызывающий так называемый «трилистник черепа». Эта черепная аномалия имеет серьезные последствия для интеллектуальных способностей и часто связана с преждевременной смертью.
Пациенты, страдающие синдромом Пфайффера II типа, представляют полную описанную выше клиническую картину последствий краниосиностоза.

ТИП III
Синдром Пфайффера III типа оказывает такое же влияние на своих носителей, как и синдром Пфайффера II типа, за исключением «черепа-трилистника».
Люди с синдромом Пфайффера типа III не имеют большой продолжительности жизни.
Аномалии больших пальцев рук и ног.
Если это особенно серьезно, аномалии, затрагивающие большие пальцы рук и ног, могут серьезно подорвать функциональные возможности рук и ног, вызывая проблемы с захватом предметов и / или ходьбой.
Вы знали, что ...
Медиальное отклонение, затрагивающее большие пальцы рук и ног у пациентов с синдромом Пфайффера, является примером варусного варуса. Точнее, врачи говорят о варусном суставе большого пальца из-за медиального отклонения большого пальца и варусном пальце из-за медиального отклонения большого пальца стопы.

Брахидактилия
При синдроме Пфайффера брахидактилия является довольно распространенной аномалией, которая может поражать только несколько пальцев или весь пальцевый комплекс рук и / или ног.
Проблема брахидактилии наблюдается во всех типологических вариантах, хотя и с разной частотой.
Синдактилия
При синдроме Пфайффера синдактилия представляет собой довольно частую «аномалию (реже, чем брахидактилия), которая может иметь различные коннотации (она может быть неполной, полной, сложной и т. Д.).
Проблема брахидактилии наблюдается во всех типологических версиях синдрома Пфайффера, хотя и с разными рецидивами.
Костный анкилоз
Синдром Пфайффера связан, прежде всего, с костным анкилозом локтя, хотя на самом деле он может вызвать ту же проблему для любого крупного сустава в организме человека.
Костный анкилоз - проблема, обнаруживаемая только при наиболее тяжелых типологических версиях синдрома Пфайффера (особенно при типе II).
Нарушения со стороны дыхательных путей
Возможные аномалии в дыхательных путях, вызванные синдромом Пфайффера, таковы, что вызывают респираторные проблемы с серьезными последствиями для общего состояния здоровья пациента (мозг страдает больше всего).
Как и при костном анкилозе, указанные выше аномалии наблюдаются только в более тяжелых типологических вариантах (в частности, тип II).
Когда можно обнаружить синдром Пфайффера?
Обычно черепные и цифровые аномалии, вызванные синдромом Пфайффера, очевидны при рождении, поэтому диагностика и планирование лечения выполняются незамедлительно.
к голове (рентгенография головы, КТ головы и / или МРТ головы) и кистям и стопам; наконец, он заканчивается генетическим тестом.
Медицинский осмотр и история болезни
Физикальное обследование и анамнез по существу состоят в точной оценке симптомов, проявляемых пациентом.
В контексте синдрома Пфайффера именно на этих этапах диагностического процесса врач выявляет краниостеноз и аномалии, поражающие большие пальцы рук и ног, и на основе других присутствующих симптомов выдвигает гипотезу о текущем типологическом варианте.
Рентгенологические исследования головы и пальцев рук и ног
В контексте синдрома Пфайффера
- Рентгенологическое исследование головы используется врачом для подтверждения наличия раннего сращения черепных швов и оценки степени тяжести черепно-мозговых аномалий.
- С другой стороны, радиологические исследования необходимы для исследования степени варуса и «возможной брахидактилии и / или» возможной синдактилии.
Генетический тест
Это анализ ДНК, направленный на обнаружение мутаций в критических генах.
В контексте синдрома Пфайффера он представляет собой подтверждающий диагностический тест, поскольку позволяет выявить мутацию FGFR2 и / или FGFR1.
Генетический тест - это также тест, который позволяет установить тип присутствующего синдрома Пфайффера.